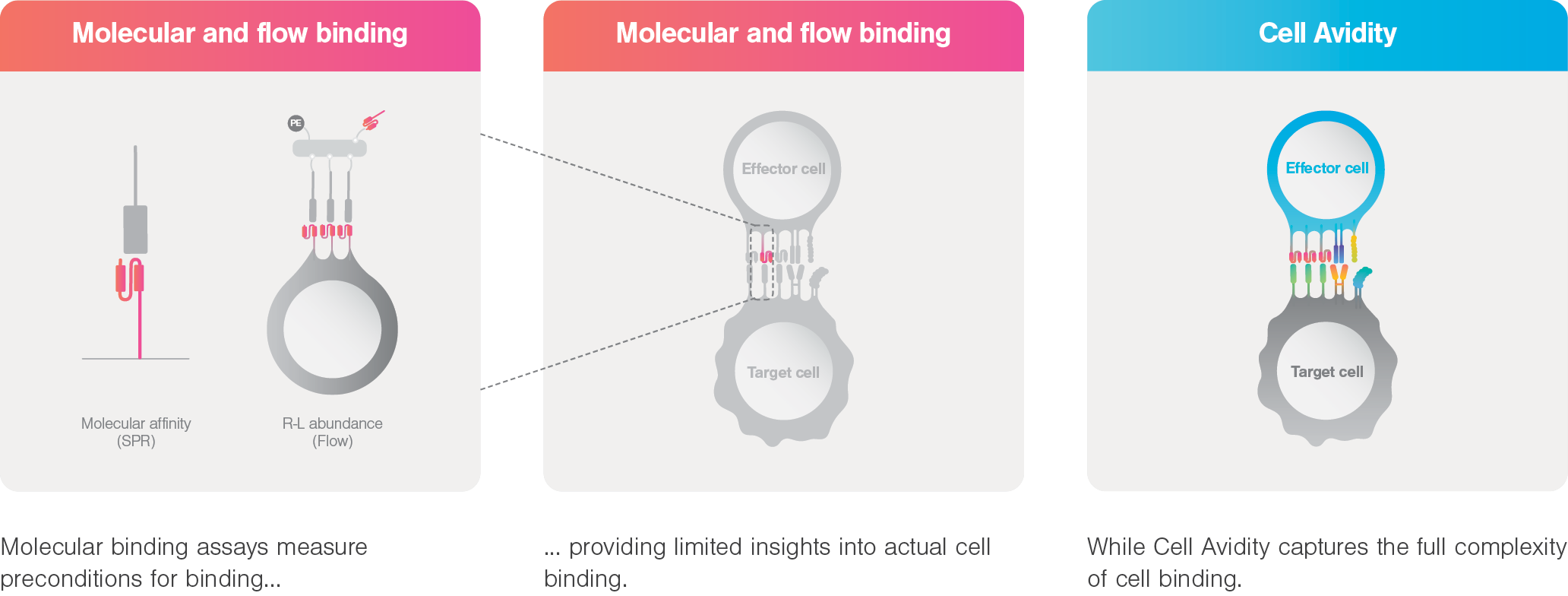
Cell Avidity
Revolutionize binding for the future of cell & antibody therapeutics
Imagine measuring the combined strength of cell-cell / cell-protein binding, generating direct, physiologically relevant measurements of binding in its full, dynamic complexity. To supportprogress in immunotherapy, we need to measure binding the way it happens—in real life, in real cells. This is Cell Avidity.
Select potent candidates, faster
Balance efficacy and safety
Accelerate your path to clinic
The challenge
There are no simple answers to immuno-oncology’s greatest challenges
Many immunotherapies adapt to solve the field’s most pressing issues (an intricate balance between persistence, potency, and safety) by integrating multiple signaling mechanisms and engaging with more than one target in parallel. With that trend, the binding mechanisms between binder and target also complexify. Yet, most binding assays haven’t kept up.

A solution for tomorrow
Cell Avidity, the missing link
By measuring the combined strength of cell-cell / cell-protein binding with controlled forces, Cell Avidity generates direct, physiologically relevant measurements of binding in its full, dynamic complexity. It reveals the mechanisms of action, facilitates rational design choices and selects the right candidates fast and early, ultimately improving therapeutic outcomes.
Customer testimonials
How Cell Avidity empowers pioneers of immuno-oncology

Mark is a global pioneer in translational cell and gene therapies
"Our study identified cell avidity as a key predictor of in vitro function, offering a pathway for rapidly developing more effective NK cell-based immunotherapies. Cell Avidity measurements [...] provide key information that can accelerate immunotherapy development by accurately predicting in vivo and clinical efficacy."
Mark Lowdell, PhD
Chief Scientific Officer
Inmune Bio

Marcela develops CAR T cell therapies to treat cancer
"Interestingly, of the pre-clinical assays we used to compare CAR constructs, only [Cell] Avidity correlated with in vivo results."
Marcela Maus, MD, PhD
Director of Cellular Immunotherapy
Massachusetts General Hospital

Peter's team explores how to create better cell-based therapies
"[Tuning Cell Avidity is] a novel method for synaptic enhancement of CAR-NKs, [which] leads to increased cellular avidity, improved function, tumor clearance and survival in in vivo solid tumor models. There’s something very elegant about the solution. Avidion makes it exceptionally straightforward."
Peter Chockley, PhD
Assistant Professor
The Ohio State University College of Medicine

Lydia's team studies how immune system dysfunction drives myeloma progression and develops rational immunotherapeutic strategies
"In our exploration of the reasons underpinning suboptimal clinical responses in the AUTO2 trial, Cell Avidity allowed us to establish poor tumor binding (avidity) by cell-bound APRIL compared to competitor CARs as a likely explanation."
Lydia Lee, PhD
Clinical Senior Research Fellow
University College London
Workflow
Intuitive, from start to finish
Measure the overall strength of interaction between cells, or between proteins and cells.
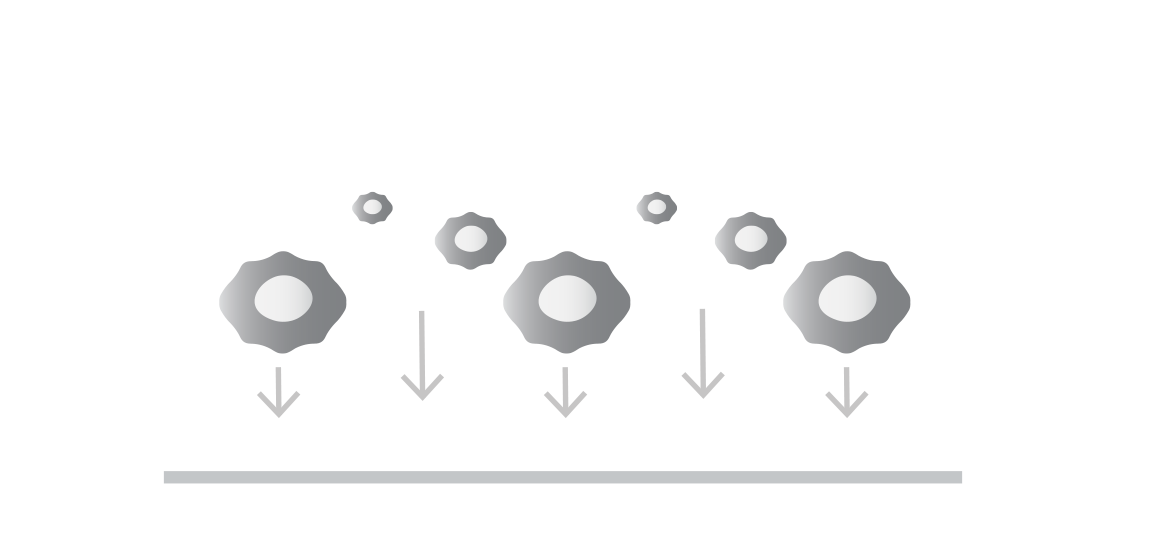
Seed your target cells
Target cells (adherent or non-adherent) are generally of two types:
Tumor cells to assess potency or antigen sensitivity
Healthy cells to assess on-target off-tumor toxicity
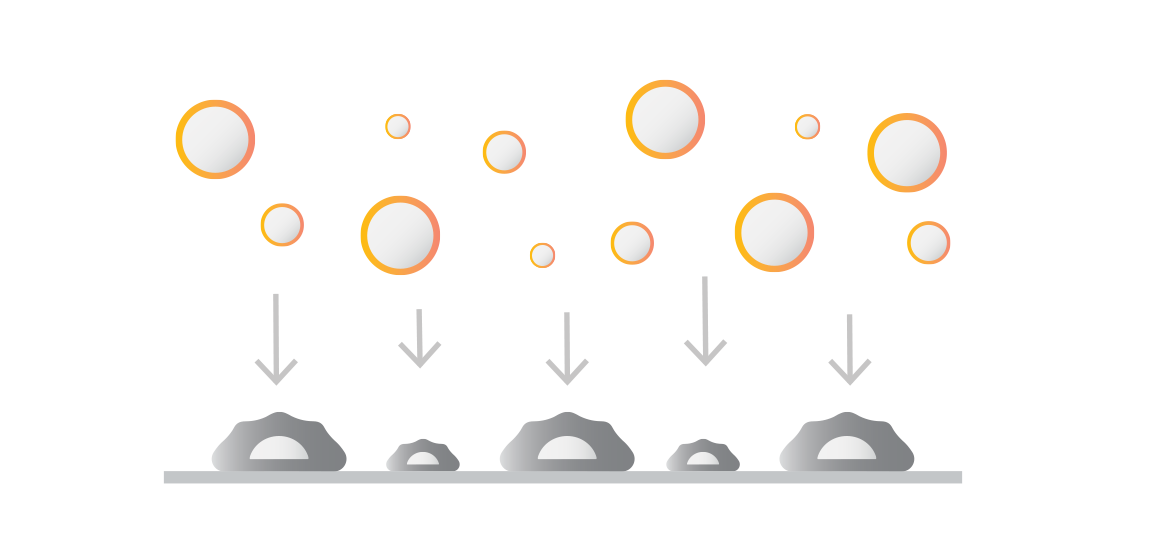
Introduce your effector cells or molecules
Introduction of labeled binders to the uniform monolayer. These can include:
Therapeutic effector cell products like CAR T, TCR T, NK.
Effector cells with compound drugs such as cell engagers, small molecule inhibitors or modified environmental factors.
Fluorescent beads coated with antibodies or other proteins.
Controlled force application & fluorescent imaging
Controlled force applied to the bound cells probes the strength of interactions between labeled and target populations. Fluorescence microscopy captures the number of bound cells before and after force application. The percentage of bound cells after force application indicates the population’s Cell Avidity.

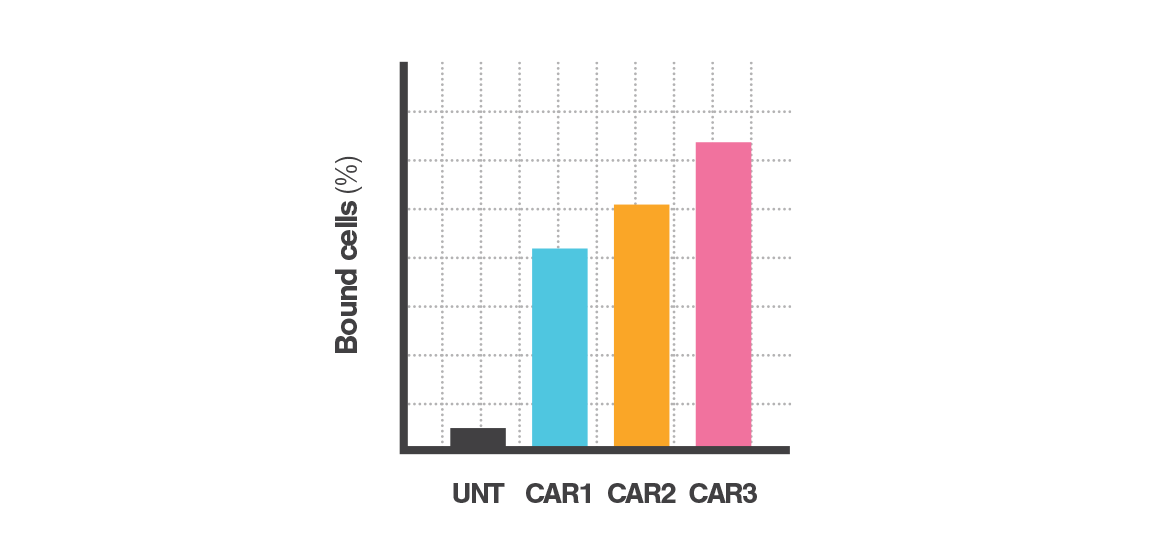
Instant data acquisition
Cell Avidity quantifies cell binding with hundreds of cells per measurement, revealing percentage (%) of cells bound, or percentage (%) of antibody-coated beads bound.
Applications
Screen cell lines, cell engagers, and antibodies, faster and better.
A Swiss army knife for next generation binding, Cell Avidity can be used to tackle a wide variety of challenges.
Cell Therapy
Steer receptor binding to overcome cell therapy exhaustion, improve potency, minimize OTOT, and refine binding sensitivity to overcome immune escape.
Featuring case studies from:



Cell Engagers
Determine avidity EC50 and binding kinetics of T cell engagers to quantify trimer formation across multiple dimensions in the cell-cell context.
Featuring case studies from:
Antibody Therapy
Elucidate binding effects of antibodies within the 2D membrane-constrained cellular environment, taking into account the effects of epitope, steric hindrance, and multivalency.
Featuring case studies from:

Tackle more than affinity ever can
Utilize Cell Avidity in a wide variety of applications.
Binding potency
Avidity ranking & cell characterization
Binding sensitivity
Titration assay for CE, Peptide or Ab
Binding specificity
Monolayer safety screening
Applicable to a variety of use-cases
Utilize Cell Avidity in a wide variety of applications.
Binder screening and optimization
Optimize binding to overcome exhaustion, improve potency, minimize OTOT and refine sensitivity to overcome immune escape.
Understand the impact of different antibody formats and fine-tune Cell Avidity to select candidates based on multi-domain binding measured in a single assay.
Effector functional modification
Measure how functional modifications in effector cells impact binding. Intracellularly driven differences in Cell Avidity can inform on potency and persistence.
Reveal the mechanism of action in therapy designs that change signaling sensitivity, activate cytokine or chemokine receptors, or reduce checkpoint receptors.
Tumor microenvironment response
Measure the tumor microenvironment’s impact on binding by measuring Cell Avidity in the presence of tumor or TME modifying compounds.
Get insights into immune trafficking, effects of environment-modulating compounds, hypoxia effects and more.
Donor selection and quality control
Measure the combined effect of donor/host interactome or assess the combined effect of cell fitness, receptor expression, and population purity.
Quantify the degree of (mis)matching between donor and host or assess product quality across multiple key product attributes.
Industry
Unlock faster R&D cycles
Integrating Cell Avidity in the drug development workflow leads to faster identification of the most potent, safe and sensitive drug candidate by reducing the required number of design iterations or eliminating candidates from in vivo preclinical studies which ultimately bind ineffectively in a cellular context.
Early in the pipeline - Select the right candidates fast
Quickly rank hundreds to thousands of therapeutic candidates to get insights into sensitivity and safety. Incorporate Cell Avidity early in the discovery workflow helps ensure that only candidates with the most promising binding characteristics progress.
Late-stage - Deep candidate characterization to drive clinical selection
Carry out deep functional characterization of select final candidates. Reveal differences in binding dynamics and target engagement to confidently select the optimal therapeutics for clinical development.
Solutions
Avidion
The next generation Cell Avidity platform
Ideal for cell therapy candidate screening and large characterization studies. Run up to 4 disposable 48-well cartridges a day for a total of 192 measurements with <80 min. hands-on time.


Avidigo
White glove Cell Avidity services
Full-service contract research from experimental design to data report based on Cell Avidity measurements at high throughput.

z-Movi
For small sized Cell Avidity studies
A fast and simple solution for single-sample Cell Avidity experiments. Run up to 20 measurements per day.
Key content
Download the brochure
Get the complete picture and specifications

Cell Avidity Technology Brochure
Brochure
The latest all-in-one overview of our Cell Avidity technology.
Technical note:
Filter CA shows 3 items
There is a hidden collection to add multiple authors to this publication.
This section is hidden when emtpy.
To keep everything visible here, that is being done outside the webflow designer from within Slater.
Filter CA shows 3 items
There is a hidden collection to add multiple authors to this publication.
This section is hidden when emtpy.
To keep everything visible here, that is being done outside the webflow designer from within Slater.
Next-generation LMP2A-targeting TCR-recombinant T cells with inducible IL-18 expression to treat EBV-associated malignancies
Bonifacius, A. et al.
2026
Molecular Therapy Oncology
This is some text inside of a div block.
Sialylated CD43 forms a glyco-immune barrier that restrains antileukemic immunity
Chung, J. et al.
2026
Science
This is some text inside of a div block.

RASA2 deletion rescues immune synapse dysfunction, enhancing CAR T cell efficacy against DMGs
Ibanez-Vega, J. et al.
2026
JITC
This is some text inside of a div block.
Adopting Cell Avidity
Let’s begin your LUMICKS journey
Our experts are ready to learn about your research challenges and see where our technologies can bring value.
Demonstrating value
Our application scientists can help create interest among potential users through organizing different events such as seminars, workshops, and demonstrations, as well as meet with stakeholders individually to understand and help solve their needs.
Avidigo services
At our Amsterdam headquarters, our internal Avidigo service team supports academic and industry clients looking to integrate Cell Avidity into their workflows and pipeline. From experimental design to data analysis and interpretation, we support you all the way.
Grant & tender support
Throughout our history we have supported multiple successful grants across a broad spectrum of funding and users involved. Our application scientists are experienced in highlighting the unique value of Cell Avidity and its solutions.